Несовершенный
остеогенез, osteogenesis imperfecta, несовершенное костеобразование,
болезнь хрустального человека, ломкость костей
Несовершенный остеогенез, osteogenesis imperfecta, несовершенное костеобразование, болезнь хрустального человека
Несовершенный остеогенез Несовершенный остеогенез (osteogenesis imperfecta) —
врожденная ломкость костей. Это сложное заболевание костей и некоторых
соединительнотканных структур, имеющее широчайший диапазон изменений,
известно с глубокой древности как заболевание с выраженной клинической
картиной и различными формами, передающееся по наследству. Первые
упоминания о нем появились в XVII в. В конце XVIII в., т.е. 200 лет
назад, Olaus Jacob Ekmann описал НО у членов одной семьи, N. Ekroth
(1788) сообщил о заболевании, которое в четырех семьях передавалось
детям, и назвал его osteomalacia congenita. Axmann (1831) не только
описал ломкость костей у себя и брата, но и, очевидно, первый отметил
такой важный симптом, как наличие голубых склер.
Lobstein (1833) описал ломкость костей у больных различного возраста.
По данным Vrolik (1849), переломы у детей происходили или еще
внутриутробно, или вскоре после рождения. Е. Looser (1906) описал эти
две формы как osteogenesis imperfecta congenita und tarda.
Изучением заболевания занимались многие врачи, описавшие более 20 различных симптомов, из которых основными являются:
изменения в строении скелета и легко наступающие переломы, часто
небольшой рост; голубые склеры; опаловидный дентин (dentinogenesis
imperfecta); прогрессирующая деформация позвоночника, грудной клетки,
черепа и длинных трубчатых костей; тугоухость по проводниковому типу;
гиперэкстензия в суставах и их деформация; изменения со стороны сердца и
крупных сосудов, носовые кровотечения и др.
Работами последних лет показано, что несовершенный остеогенез является
гетерогенным наследственным заболеванием генетической природы,
поражающим соединительную ткань и выражающимся остеопенией и
вышеперечисленными клиническими признаками.
Вместо двух форм, или типов, в настоящее время по предложенной в 1979
г. D.O. Sillence классификации несовершенный остеогенез с учетом
клинических, рентгенологических и коллагеновых протеин-генных
молекулярных изменений подразделяют на 4 типа.
Тип I — слабовыраженная форма, доминантно-наследственный несовершенный остеогенез с ломкостью костей и голубыми склерами.
Тип II — перинатально-летальный.
Тип III — прогрессирующее деформирование скелета.
Тип IV — доминантный с нормальными склерами и нерезко выраженными деформациями.
Общие проявления. Термином несовершенный остеогенез обозначают наследственные аномалии, обусловливающие хрупкость костей.
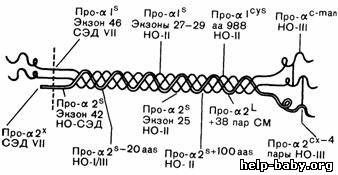
Рис.319-2.Приблизительная локализация мутаций в структуре проколлагена I типа.
Римскими цифрами обозначен конкретный тип синдрома Элерса —Данло
(СЭД) или несовершенного остеогенеза (НО), обсуждаемых в тексте. Экзоны,
в которых происходят специфические делеции, пронумерованы в направлении
от 3- к 5-концу гена. Другие делеции обозначены примерным числом
утраченных аминокислот; аа 988 означает, что остаток глицина в положении
988 a1-цепизамещен цистеином. Как сообщалось в тексте, мутация про-a21означает
вставку 38 пар оснований в дополнительную последовательность и
обнаружена у больных с атипичным синдромом Марфана (СМ); про-a2^looaasозначает делецию примерно 100 аминокислот при a-варианте несовершенного остеогенеза II типа.
Про-a^—мутация, ведущая к укорочению npo-al-цепи; про-(^—мутация, ведущая к укорочению ^1ро-а2-цепи; про-а!^5—мутация, ведущая к появлению цистеинового остатка;пpo-a:~ma"—мутация,
ведущая к избыточному содержанию маннозы в одной или обеих про-а-цепях;
про-а2"— неизвестная структурная мутация, препятствующая расщеплению
цепи N-протеиназой; про-а21— мутация, ведущая к удлинению про-а2-цепи; про-с^0"—
мутация, меняющая структуру С-концевого пропептида про-а2-цепи
(модифицировано и воспроизведено с разрешения из ProckopandKivirikko). Мальчик в возрасте 21 мес с несовершенным остеогенезом III типа.
Ребенок страдает множественными переломами рук и ног. Он гомозиготен по
делеции 4 пар оснований в генах про-а2(1)-цепей, что привело к изменению
последовательности последних 33 аминокислот в этих белках. В связи с
этим про-а2(1)-цепи не сомкнулись с про-а1 (I) -цепями, и единственной
формой проколлагенов I типа оказались тримеры про-al (I) -цепей, в
которых С-концевые участки остались нескрученными (воспроизве- навливают
путем исключения других наследственных дефектов или влияний факторов
окружающей среды, вызывающих остеопению или остеопороз, и выявления
последствий мутации в нескольких видах соединительной ткани. Повышенная
ломкость костей сопровождается обычно такими признаками, как голубой
цвет склер, глухота, нарушение прорезывания зубов. Эти признаки могут
определяться по отдельности или вместе (табл.319-2). Для того чтобы
установить диагноз в раннем детстве, достаточно выявить сочетание
голубого цвета склер и переломов. Точно так же достаточно определить
сочетание переломов с характерными аномалиями зубов (несовершенный
дентиногенез). Некоторые специалисты диагностическое значение придают
сочетанию ломкости костей с наступившей рано глухотой у больного или
членов его семьи, тогда как другие ставят диагноз только на основании
хрупкости костей, которую нельзя связать с внешними факторами (такие,
как малая физическая активность или сниженное питание) или с другими
наследственными синдромами, например дисплазиями скелета (табл.319-3).
Поскольку у некоторых членов семей переломов не бывает до наступления
постменопаузы, легкие формы болезни могут быть неотличимы от
постменопаузального остеопороза. Некоторые лица с остеопорозом могут
быть гетерозиготными носителями генных дефектов, вызывающих у гомозигот
несовершенный остеогенез. В связи с этим целесообразно отнести
постменопаузальный остеопороз в спектр тех же болезней, к которым
относится несовершенный остеогенез.Для классификации несовершенного остеогенеза пользуются
классификацией, предложенной Sillence (см. табл.319-2). Тип I
встречается с частотой примерно 1:30 000. Он представляет собой легкую
или средней тяжести болезнь, наследуемую как аутосомный доминантный
признак в сочетании с голубыми склерами. Наиболее тяжело протекает
болезнь II типа. Типы III и IV по тяжести занимают промежуточное
положение между типами I и II.
Аномалии скелета. При I
типе болезни ломкость костей может быть столы выраженной, что
ограничивает физическую активность больного, или столь незначительной,
что больной вообще не испытывает никаких неудобств. При II типе кости и
другие виды соединительной ткани настолько хрупки, что смерть наступает
еще в утробном периоде, в родах или в первые несколько недель после
рождения ребенка. При болезни III и IV типов множественные переломы,
возникающие даже при минимальных физических воздействиях, могут привести
к остановке роста и костным уродствам. У многих больных переломы
особенно часто возникают в детстве; после периода пубертата их частота
уменьшается, а при беременности и после наступления менопаузы вновь
увеличивается. Резкий кифосколиоз может быть причиной нарушений дыхания и
предрасполагать к легочным инфекциям. Плотность костей снижена, но
относительно специфических морфологических нарушений мнения расходятся.
Общее впечатление таково, что заживление переломов происходит нормально.
У некоторых больных со сравнительно легкой симптоматикой череп имеет
множество вмятин, по-видимому, из-за небольших очажков оссификации
Глазные симптомы. Цвет склер варьирует от нормального до
слегка голубоватого или от синевато-серого до ярко-голубого. Голубизна
обусловлена истончением или прозрачностью коллагеновых волокон склеры,
через которые просвечивает сосудистая оболочка глаза. У ряда больных
выявляют и другие глазные симптомы. В некоторых семьях голубые склеры
могут быть наследственным признаком без всякого увеличения хрупкости
костей.
Несовершенный дентиногенез. Эмаль твердой зубной пластинки
относительно нормальна, но зубы имеют янтарный, желтовато-коричневый или
полупрозрачный голубовато-серый цвет из-за неправильного отложения
дентина. Молочные зубы обычно мельче нормальных, а постоянные заострены и
как бы имеют основание. Точно такие же аномалии зубов могут
наследоваться независимо от несовершенного остеогенеза.
Глухота. В возрасте после 10 лет или позднее развивается
глухота. Она обусловлена нарушением прохождения колебаний через среднее
ухо на уровне основания стремени. При гистологическом исследовании
обнаруживают недостаточную оссификацию, персистенцию хрящевых участков,
которые в норме оссифицируются, и полоски скопления кальция.
Сопутствующие проявления. У многих больных и у членов многих
семей выявляют аномалии и в других видах соединительной ткани. В
некоторых случаях отмечают изменения кожи и суставов, неотличимые, от
таковых при синдроме Элерса —Данло (см. далее). У небольшого числа
больных выявляют нарушение функции сердечно-сосудистой системы, например
регургитацию аортальных клапанов, пролабирование митральных, митральную
недостаточность и хрупкость стенок крупных кровеносных сосудов. Могут
иметь место гиперметаболизм с повышением уровня тироксина в сыворотке,
гипертермия и чрезмерная потливость. При легких формах болезни
сопутствующие симптомы могут выступать на первый план.
Способ наследования. Тип I болезни наследуется как аутосомный
доминантный признак с непостоянной экспрессией, так что он может
проявляться через поколение. При летальном варианте II типа наследование
может быть аутосомным рецессивным, но в нескольких случаях II типа с
выясненным генетическим дефектом имелись новые мутации. Способ
наследования — основной критерий разграничения III и IV типов (см.
табл.319-2), но отличить рецессивно наследуемую форму от новой
аутосомной доминантной мутации иногда очень трудно.
Молекулярные дефекты. Поскольку большинство тканей при
несовершенном остеогенезе богато коллагеном I типа, считают, что многие
его формы связаны с мутациями структурных генов этого белка, генов,
определяющих его посттрансляционный процессинг, или генов, регулирующих
его экспрессию. В настоящее время выяснены мутации генов проколлагена I
типа при четырех вариантах II типа несовершенного остеогенеза. Один
вариант характеризовался делецией в одном из аллелей гена про-al (I)
(рис.319-4). Она распространялась на три экзона, но не препятствовала
транскрипции гена. В результате про-al (I)-цепь оказалась на 84
аминокислоты короче, чем в норме. Эта мутация была летальной, поскольку
укороченная про-al (I)-цепь связывалась с нормальной про-al (I)- и
про-а2(1)-цепями (см. рис.319-4). Укорочение про-al (I)-цепи
препятствовало скручиванию молекул в тройную спираль. В связи с этим
большая часть проколлагеновых молекул оставалась нескрученной и быстро
распадалась в процессе, называемом самоубийством белка, или негативной
комплементарностью (см. рис.319-4). При втором летальном варианте
болезни II типа мутация привела к синтезу такой про-а2(1)-цепи, которая
была примерно на 20 аминокислот короче по сравнению с нормой. Второй
аллель не функционировал, поэтому все про-а2-цепи оказались
укороченными. При третьем варианте II типа мутационная делеция в аллеле
про-а2(1)-цепи укоротила синтезируемую про-а2-цепь примерно на 100
аминокислот. При четвертом варианте II типа происходило замещение одного
основания, что привело к появлению в a1(1)-цепи остатка цистеина вместо
глицина и тем самым к разрыву трехспиральной конформации белка.
Мутации генов проколлагена I типа выяснены также при двух вариантах
болезни III типа. При одном из них была определена делеция четырех пар
оснований, что изменило последовательность последних 33 аминокислот в
про-а2(1)-цепи. Больной был гомозиготен по этому дефекту, и ни одна из
про-а2(1)-цепей не включалась в молекулы проколлагена. Вместо этого
проколлаген I типа состоял из тримера про-al (I)-цепей. Этот тример имел
трехспиральную конфигурацию, но был нестабильным. Родители больного,
находившиеся друг с другом в троюродном родстве, были гетерозиготами по
той же мутации и уже в возрасте 30 лет страдали остеопорозом. При другом
варианте III типа структурные изменения в С-концевом пропептиде
обусловили увеличение количества в нем маннозы. У больного с некоторыми
симптомами болезни I типа и другими, типичными для болезни II типа,
про-а2(1)-цепи были укорочены примерно на 100 аминокислот.
На основании этих данных можно сделать ряд обобщений в отношении
мутаций генов коллагена. Одно из них сводится к тому, что мутация,
ведущая к синтезу аномального белка, может быть более вредной, чем
нефункционирующий аллель. Второе заключается в том, что мутации,
обусловливающие укорочение полипептидных цепей, могут быть более
частыми, чем другие. Однако у большинства больных молекулярные дефекты
не идентифицированы. У многих из них могли иметь место мутации
сплайсинга РНК или мутации по единичным основаниям, которые трудно
обнаружить в столь крупных генах, как ген проколлагена I типа. Ряд
вариантов несовершенного остеогенеза мог бы обусловливаться мутациями
других генов, экспрессия которых необходима для сборки и сохранения
структуры костей и других видов соединительной ткани.
Диагностика. В отсутствие кардинальных признаков болезни
диагноз установить трудно, и многие случаи, вероятно, остаются
недиагностированными. Следует учитывать возможность других
патологических состояний, сопровождающихся хрупкостью костей в
младенчестве и детстве (см. табл.319-3). У 1/3 больных при электрофорезе
проколлагена I типа (синтезируемый фибробластами кожи в культуре) в
полиакриламидном геле можно обнаружить аномальную про-a-цепь. В
большинстве случаев изменение подвижности отражает посттрансляционную
модификацию и не позволяет определить точную природу мутации или тип
болезни.
Лечение. Убедительные данные о возможности эффективного
лечения отсутствуют. При легкой форме после уменьшения частоты переломов
в возрасте 15— 20 лет больные могут и не нуждаться в лечении, но во
время беременности или после наступления менопаузы, когда частота
переломов снова увеличивается, к ним требуется особое внимание. При
более тяжелых формах детям необходимы широкая программа физиотерапии,
хирургическое лечение при переломах и. деформациях скелета,
профессиональное обучение и эмоциональная поддержка как больному, так и
его родителям. У многих больных интеллект достаточно развит, и они,
несмотря на выраженные деформации, делают успешную карьеру.
Целесообразно использовать программу поддержания позы, разработанную
Bleck. При многих переломах лишь минимально смещаются кости и происходит
некоторый отек мягких тканей, поэтому требуется лишь слабое вытяжение в
течение 1—2 нед с последующим наложением легкой шины. При
малоболезненных переломах необходимо рано начинать физиотерапию. В
отношении целесообразности коррекции деформаций конечностей с помощью
стального гвоздя, помещаемого в длинные кости, мнения противоречивы.
Оправданием этой процедуры может служить то обстоятельство, что
коррекция деформаций в детстве дает возможность взрослым больным
нормально ходить.
Генетическое консультирование при II, III и IV типах болезни
затруднено из-за неясности способа наследования. С помощью рентгене- и
эхографии несовершенный остеогенез удавалось диагностировать у плода уже
на 20-й неделе беременности. В тех немногих семьях, где точно выяснен
генный дефект, для пренатальной диагностики можно было бы производить
анализ ДНК в соответствующих лабораториях. Для генов проколлагена I типа
идентифицирован полиморфизм длины рестрикционных фрагментов, и этот
подход можно было бы использовать для пренатальной диагностики. Культура
клеток амниотической жидкости синтезирует коллаген, но применять эти
культуры для выявления мутаций представляется нереальным. |













